- COORDINATION (chimie) - Chimie de coordination
- COORDINATION (chimie) - Chimie de coordinationLes cations n’existent pratiquement jamais seuls; dans tous leurs composés, ils sont environnés d’anions ou de molécules neutres. On appelle ligands (ou coordinats) les groupements assurant l’entourage immédiat d’un cation. La discipline qui traite des propriétés de ces associations entre cations et ligands est la chimie de coordination.Aucune démarcation très nette n’existe entre la chimie de coordination et la chimie des molécules covalentes organiques ou inorganiques, d’une part, et la chimie des solides tridimensionnels homodesmiques ou hétérodesmiques, d’autre part. Mais l’essentiel de la discipline concerne les cations des éléments de transition, c’est-à-dire des éléments métalliques qui possèdent une couche d, ou bien f s’il s’agit d’une terre rare, incomplète.Lorsque l’association entre un (ou plusieurs) cation métallique (et par extension atome ou anion) et les ligands est une entité indépendante, molécule, anion ou cation, on retrouve aisément la notion de complexe de coordination , conforme à la description classique que les travaux effectués par S. M. Jørgensen (1837-1914) et par A. Werner (1866-1919, prix Nobel de chimie en 1913), conduisirent à dégager et que les théories de Kossel, de Lewis, de Bethe, de Van Vleck, de Pauling, de Mulliken et Slater permirent de préciser.L’objet même de la chimie de coordination, à savoir l’ion (ou l’atome) métallique complexé qui fait de cette discipline une branche maîtresse de la chimie minérale, lui a, au cours du temps, ménagé des alliances diverses. On peut rappeler l’une des plus intimes, avec la chimie analytique, qualitative ou quantitative (cf. COMPLEXES [chimie]) ou encore celle, nécessaire, avec la chimie physique. Cependant, on doit reconnaître que le fait le plus essentiel, depuis le début des années soixante, réside en un déploiement remarquable de la chimie de coordination vers la chimie organique sous forme de ce qu’il est convenu d’appeler la chimie organométallique des éléments de transition. C’est dans ce cadre que la chimie de coordination s’identifie partiellement à la chimie moléculaire de ces éléments.L’importance de ce phénomène a été soulignée par l’attribution en 1963 du prix Nobel de chimie à K. W. Ziegler (R.F.A.) et à G. Natta (Italie), pour leurs travaux sur la polymérisation stéréospécifique des oléfines catalysée par des mélanges d’halogénures de métaux de transition et d’alkylaluminium, et en 1973 à G. Wilkinson (Royaume-Uni) et à E. O. Fischer (R.F.A.) pour, en particulier, leur contribution à la chimie des métallocènes. Le premier exemple montre par ailleurs que le développement de cette interface entre la chimie de coordination et la chimie organique ne peut être dissocié de celui de la catalyse homogène par complexe.1. Les complexes de ligands size=5神-accepteursUne propriété caractéristique des éléments de transition est leur capacité à former des complexes avec un grand nombre de molécules neutres, au premier rang desquelles on peut citer l’oxyde de carbone, les isocyanures, les phosphines, arsines, etc., ainsi que diverses molécules aux orbitales 神 délocalisées, comme la pyridine, l’orthophénanthroline. Dans beaucoup de ces complexes, l’atome métallique central se trouve dans un état d’oxydation formelle soit faiblement positif, soit nul ou même négatif. Cette stabilisation des états d’oxydation faibles tient au fait que ces ligands, possédant des orbitales 神 vacantes, sont susceptibles d’accepter un transfert d’électrons depuis les orbitales pleines du métal. Ainsi la charge de l’atome métallique, même dans ses états d’oxydation très faibles, peut-elle rester sensiblement positive.Les métaux carbonylesLe plus important des ligands 神-acides est l’oxyde de carbone. Il conduit à des composés dont les plus simples sont du type M(CO)x ; les plus connus figurent dans le tableau 1 ainsi que la nature de leurs polyèdres de coordination. Dans tous les cas, ces groupements M-C-O sont linéaires et les métaux carbonyles possèdent donc une structure très symétrique.La stabilité des métaux carbonyles tient à la nature multiple de la liaison M-C-O. La figure 1 représente les recouvrements qui permettent l’établissement, d’une part de la liaison 靖 donneur-accepteur, carbonemétal, et d’autre part de la liaison 神 donneur-accepteur, métalcarbone, constituant cette double liaison.Il existe de nombreux dérivés des métaux carbonyles obtenus par substitution aux différents groupements CO d’autres ligands L, tels les phosphines, les oléfines, les composés aromatiques, etc. Dans ces composés de formule générale M(CO)x -n Ln , les diverses liaisons de coordination s’établissant entre les ligands et le métal central peuvent être décrites suivant un schéma comparable à celui donné ci-dessus. Cette situation conduit à l’existence d’un équilibre électronique entre les différents ligands qui joue un rôle important aussi bien au niveau structural qu’en ce qui concerne les réactions de ces composés.Les métaux carbonyles et leurs dérivés mononucléaires fournissent des exemples remarquables de conformité à la règle formelle de Sidgwick du nombre atomique effectif. Cette tendance est respectée par les métaux de nombre atomique impair, qui forment dès lors des complexes dinucléaires à liaison métal-métal, tels Co2(CO)8 et Mn2(CO)10. De telles structures polynucléaires sont propices à l’établissement de pontages assurés par les groupements CO entre atomes métalliques (fig. 2).À partir des métaux carbonyles, il est possible, dans de nombreux cas, d’obtenir par diverses méthodes des anions carbonylates, tels Co(CO)-4, Fe(CO)42- ou bien encore, à partir de Cr(CO)6, Cr2(CO)102- .Des composés halogénés M x (CO) y Xz sont connus pour la plupart des éléments formant des carbonyles binaires, mais aussi pour le palladium, le platine et l’or, qui n’en forment pas. De nombreux cas de structures dimères ou polymères existent; elles sont généralement le résultat d’un pontage par les ions halogénures.Les dérivés des ligands des groupes V et VI. Les hydrures complexesDe très nombreux dérivés du phosphore trivalent (PR3), de l’arsenic (AsR3) ainsi que du soufre divalent fournissent des complexes avec les métaux de transition dans divers états de valence. Une de leurs propriétés remarquables est de stabiliser des complexes à liaison métal-hydrogène ou métal-carbone, longtemps supposés instables. Dans ces composés, la situation électronique de l’hydrogène est celle d’un hydrure comme en témoignent, en particulier, les spectres de résonance magnétique nucléaire. Par ailleurs, on peut obtenir des hydrures complexes (fig. 3), notamment par protonation des complexes de ligands basiques et surtout des anions carbonylates.Les complexes de ligands à système size=5神 étenduCertaines molécules possédant notamment des atomes donneurs d’azote (structures polypyridiniques) ou de soufre (dithiolène) possèdent la propriété remarquable de former des complexes de cations métalliques pour un large éventail d’états d’oxydation. Ce phénomène est essentiellement dû à une importante délocalisation électronique sur les ligands, comme en témoignent les résultats obtenus par résonance paramagnétique électronique de dérivés de l’ 見, 見 , dipyridile, tels:2. Les composés organométalliquesLes complexes dérivés des alcènesBien que, déjà en 1830, W. C. Zeise ait pu obtenir un composé du platine et de l’éthylène, la capacité des métaux de transition à fournir des composés organométalliques n’a été vraiment reconnue qu’au cours des années cinquante. L’anion PtCl3C2H4-(fig. 4 a) en est certainement l’exemple le plus simple, à propos duquel a pu être développé un modèle de liaison (connu sous le nom de modèle de Chatt-Dewar) qui, avec ses deux composantes 靖 (C 轢M) [fig. 4 b] et 神 (M 轢C) [fig. 4 c], est très voisin de celui qui est retenu pour les liaisons M 漣 CO et M 漣 ER3 (avec E = P, As, Sb...).La chimie des composés mono-oléfiniques et polyoléfiniques est extrêmement développée, en particulier dans ses relations avec les phénomènes catalytiques. Les ligands 1,5-cyclo-octadiène et norbornadiène mettant à profit l’effet de chélation ont été très souvent utilisés.Les complexes dérivés des alcynesIl existe de nombreux complexes dans lesquels un carbure acétylénique joue essentiellement le même rôle qu’un oléfine. Dans d’autres cas, l’existence de deux systèmes 神 dégénérés conduit l’alcyne, par duplication de la situation existant avec les oléfines, à se lier à deux atomes métalliques. C’est ce qui apparaît par exemple dans les composés Co2(CO)6(RC 令 CR) : fig. 5.Dans certains cas enfin, tel celui du complexe L2Pt(PhC2Ph), la meilleure description du ligand acétylénique se rapproche de celle d’un dianion comme résultat d’une «addition oxydante» (cf. chap. 3) sur le platine.La liaison size=5靖 M-CAprès des tentatives infructueuses faites pour obtenir des composés alkylés ou arylés des métaux de transition simples (en l’absence de ligands 神-accepteurs stabilisants), il est apparu que l’apparente «instabilité» ainsi observée n’était pas le résultat d’une faible énergie de liaison M-C. Le plus souvent, il s’agit d’une labilité cinétique importante due à une possibilité de 廓-élimination (réaction inverse de l’«insertion») d’un atome d’hydrogène (cf. chap. 3).La liaison métal-carbone dite 靖 simple apparaît dans de très nombreux composés et on peut citer des cas remarquables comme Ti(CH3)4, W(CH3)6 ou (CO)5Mn(CH3). Par contre, on doit admettre l’existence de liaisons métal-carbone multiples dans des complexes où se trouvent stabilisées des formes carbéniques, ainsi (fig. 6) dans:
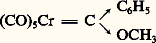 ou des formes carbyniques dans:
ou des formes carbyniques dans: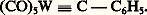 Les complexes de ligands hydrocarbonés cycliquesDans ce domaine, le ferrocène Fe(C5H5)2 est certainement remarquable à la fois par sa stabilité (il n’est pas détruit à 500 0C) et par sa structure (fig. 7). Dans le motif C5H5Fe, les cinq atomes de carbone de l’anion cyclopentadiényle sont également liés à l’atome métallique et forment une couronne possédant un axe de symétrie d’ordre cinq. Ce motif se retrouve dans de nombreux dérivés appelés parfois «composés sandwich», structuralement analogues au ferrocène, les métallocènes M(C5H5)2, avec M = Ni(II), Cr(II), Co(II), etc., mais aussi dans des dérivés pseudotétraédriques comme (C5H5)2TiCl2 ou (C5H5)2WH2. Par ailleurs, l’anion cyclopentadiényle n’est pas le seul qui puisse être lié symétriquement à un atome métallique; les espèces cycliques C3R3+, C4H42-, C6H6, C7H7+ , C8H82- et leurs dérivés qui, comme C5H5- , possèdent un nombre d’électrons susceptible de leur conférer l’aromaticité, forment également des complexes remarquables dont le prototype est le chrome dibenzène Cr(C6H6)2. On pourra noter de plus que certains anions dérivés du bore, en forme de nid , tel B9C2H11- , conduisent à des composés de type voisin.Enfin, il a été possible de construire des édifices (C5H5)3M2 dans lesquels les trois cycles cyclopentadiényle superposés emprisonnent deux à deux, entre leurs plans parallèles, chacun des deux atomes métalliques placés sur l’axe commun de symétrie de l’ensemble.3. États d’oxydation, coordinences et ligands non usuelsLa chimie de coordination a puisé une part importante de sa dynamique dans la recherche des moyens propres à stabiliser des états d’oxydation particuliers et des états de coordinences non usuelles.Une partie de ce qui précède illustre les très nombreuses possibilités de stabilisation d’états d’oxydation faibles. En ce qui concerne les états d’oxydation élevés des métaux, on sait qu’ils sont souvent stabilisés à l’état d’anions ou polyanions minéraux qu’ils forment avec les ligands 2- et F - . Ils se rencontrent également dans des complexes dérivés de macrocycles azotés [cas de Fe(IV), Mn(IV), Ni(III)...] et de ligands soufrés (dithiocarbamate, dithiolate en particulier). On attribue également à ces états d’oxydation élevés un rôle important en milieu biologique où ils forment des complexes avec certains peptides et polypeptides.L’obtention de complexes présentant des états de coordinence non usuels est facilitée par l’emploi de ligands contraignants, susceptibles d’imposer un environnement particulier à l’ion métallique. Les porphyrines, ligands tétradentates rigides imposant une structure de plan carré, ainsi que certains multidentates [tel P(C6H4N 漣 CH=NH)3, par exemple, qui impose la géométrie de prisme droit] illustrent cet aspect de la stabilisation des complexes.Certaines molécules usuelles simples, dès lors qu’il fut montré qu’elles peuvent jouer un rôle de ligand, ont conduit au développement de larges secteurs de la chimie de coordination. L’un de ceux-ci concerne les complexes de l’azote moléculaire qui jouent un rôle important – de modèles – dans notre compréhension de la fixation de l’azote atmosphérique par les plantes. On en connaît maintenant des exemples pour la plupart des métaux de transition et les propriétés observées pour certains d’entre eux, tel le complexe du molybdène et du bis(diphénylphosphine)éthane, Mo(N2)2(dppe)2, permettent de fonder des espoirs quant à la mise au point de méthodes de synthèse directe de produits azotés intéressants.Les complexes de l’oxygène moléculaire sont également très étudiés, qu’il s’agisse de composés mononucléaires symétriques, dans lesquels les deux atomes d’oxygène sont équidistants du métal, de complexes mononucléaires dans lesquels un seul des atomes d’oxygène est lié au métal, ou qu’il s’agisse enfin de composés dinucléaires dans lesquels l’oxygène sert de pont entre les atomes métalliques. Leur étude apporte des éclaircissements sur le fonctionnement des porteurs d’oxygène (hémoglobine, hémocyanine), d’une part, des oxygénases, des oxydases et des oxygénations catalytiques, d’autre part.Les complexes polynucléairesHier encore simples cas particuliers sans grande portée générale, les complexes polynucléaires des métaux de transition sont l’objet aujourd’hui d’un intérêt considérable, principalement dans le cas où ils font apparaître des liaisons entre atomes métalliques. Dans ce cas précis, on peut distinguer deux catégories de composés, à savoir:– les complexes comportant une (ou plusieurs) liaison métal-métal à deux centres métalliques identiques ou différents; les cas les plus simples de tels complexes dimétalliques sont apparentés à celui de Co2(CO)8 (fig. 2) par le fait que la liaison métal-métal y assure seule l’assemblage de deux motifs monométalliques; dans certains composés, au contraire, les ligands en pont entre les atomes métalliques (tels Cl- , OR- , SR - , PR2- ou RC2- ) viennent renforcer cet assemblage; enfin, des liaisons métal-métal multiples apparaissent pour certaines configurations électroniques relativement pauvres en électrons; on admet ainsi, dans le cas de l’octachlorodimolybdate Mo2Cl84- , dans lequel la distance Mo-Mo est très courte, l’existence d’une liaison quadruple;– les complexes contenant au moins trois atomes métalliques, identiques ou différents, souvent disposés suivant des arrangements polygonaux réguliers (fig. 8); dans ces clusters , les électrons de liaison sont délocalisés sur l’ensemble des atomes métalliques, par ailleurs dans un état d’oxydation formel souvent nul; l’ensemble n’est donc pas sans évoquer le métal massif; les métaux de transition les plus aptes à la synthèse de clusters sont ceux qui se situent sur la droite du tableau périodique; le panorama des structures offert actuellement est extrêmement varié, qu’il s’agisse de composés homo-ou même hétérométalliques, et on connaît maintenant des cas de haute nucléarité (18 atomes de platine par exemple); enfin, certains de ces clusters contiennent, insérés dans le polyèdre métallique, divers petits atomes dans des états de coordinence très inhabituels en chimie moléculaire (fig. 9).Un regain d’intérêt semble enfin se porter sur les polyanions ; ces édifices polymétalliques ne sont en fait pas des clusters dans la mesure où ils ne font pas intervenir d’interaction métal-métal significative sur le plan structural. Résultant le plus souvent de condensations réalisées à partir des ions oxo M4n - (M = VV, NbV, TaV, MoVI, WVI), ils possèdent diverses stœchiométries, tel Nb6198-, que l’on peut proposer comme exemple simple d’isopolyanion, ou (Co2W1242)8- , exemple d’hétéropolyanion. Dans la plupart des cas, ces ions sont formés d’octaèdres M6 partageant sommet et arêtes, à l’exclusion des faces (fig. 10). On peut noter enfin, pour certains d’entre eux, des possibilités de réduction multiélectronique conduisant à des composés à valence mixte.4. Développements récents des méthodes d’études expérimentales et théoriquesLes synthèsesUne grande partie des progrès de la chimie des complexes, en particulier dans le domaine des composés organométalliques, repose sur le développement de méthodes synthétiques fines, tel l’emploi des milieux rigoureusement inertes et celui des basses températures.Les méthodes de séparation sont également d’une grande importance, en particulier les méthodes de chromatographie sur plaque et de chromatographie liquide sous pression. Une mention particulière doit être faite des méthodes de synthèse électrochimique, photochimique, et surtout de la vaporisation d’atomes métalliques. Cette dernière technique, qui consiste à faire réagir directement la vapeur métallique avec des substrats, des ligands divers, a permis de préparer des espèces longtemps inaccessibles, par exemple Ti(C6H6)2.Analyse structuraleLa généralisation des méthodes d’analyse cristallographique, au moyen notamment des diffractomètres automatiques, rend possible la détermination directe des structures moléculaires, qui constitue dans certains domaines des informations strictement irremplaçables (par exemple dans le domaine des clusters). Des techniques encore plus fines combinant la diffraction des rayons X et celle des neutrons autorisent la détermination directe des densités électroniques, en particulier au sein même des liaisons chimiques.Une remarque s’impose qui concerne l’utilisation des méthodes physiques d’investigation. La chimie de coordination est en effet pour une large part – ce qui la différencie largement de la chimie organique classique dans le domaine des déterminations structurales – une chimie des édifices moléculaires de haute symétrie centrée. Et cela confère un rôle essentiel à la théorie des groupes dont l’usage s’est donc imposé aussi bien en spectrographie infrarouge et Raman qu’en spectroscopie électronique ou de résonance magnétique nucléaire ou électronique.Les méthodes d’approche théoriqueLes approches qualitatives, issues de ce qu’il fut convenu d’appeler «théorie des liaisons de valence dirigée», «théorie du champ cristallin» puis «théorie des orbitales moléculaires», s’intègrent maintenant complètement dans un modèle plus large, celui du «champ des ligands» dont elles représentent en quelque sorte des étapes successives d’approximation. Cela résulte directement du fait que chacune de ces approches devait tenir compte de la symétrie de la molécule qui est en fait leur facteur commun. On peut noter également le développement du modèle dit de recouvrement angulaire, qui fournit une évaluation commode de l’énergie de stabilisation liée à la disposition des ligands autour du métal suivant une certaine géométrie. L’effort est maintenant porté sur la mise au point de méthodes de calcul applicables aux atomes lourds des seconde et troisième séries de métaux de transition.5. Réactivité et catalyseLe perfectionnement des concepts classiquesLes réactions de substitution nucléophile du premier et du deuxième ordre S1 et S2 sont étudiées jusqu’au stade le plus intime de leur mécanisme. De plus, des acquisitions importantes dans le domaine de la réactivité ont résulté de l’introduction de la notion de non-rigidité des structures complexes elles-mêmes.La pseudo-rotation dite de Berry permet d’expliquer l’équivalence apparente des ligands axiaux et équatoriaux d’un complexe de structure bipyramidale triangulaire par une déformation légère conduisant à un intermédiaire de structure pyramidale à base carrée [cf. DYNAMIQUE MOLÉCULAIRE].Les réactions fondamentales des complexes organométalliquesCertaines réactions des complexes ont suscité tout particulièrement l’intérêt dans la mesure où elles mettent en évidence la capacité du métal central à modifier profondément l’état structural des réactifs pénétrant la sphère de coordination. Le tableau 2 présente quelques-unes de ces réactions.Ces processus, qui font souvent jouer un rôle de gabarit au métal central du complexe, sont parties intégrantes des mécanismes des réactions catalysées par les complexes des métaux de transition (cf. CATALYSE – catalyse homogène).6. PerspectivesIl est certain que la catalyse homogène organique se place maintenant au tout premier rang des applications de la chimie de coordination (cf. CATALYSE – catalyse homogène). Le domaine des complexes de coordination d’intérêt biologique (on pensera en premier lieu aux chlorophylles et à l’hémoglobine) est également aujourd’hui une puissante motivation pour les spécialistes de la chimie de coordination. De plus, la découverte récente des propriétés antitumorales de certains complexes, en particulier de complexes dérivés du platine, semble marquer le renouveau d’une pharmacopée plus «minérale». Ces domaines relèvent de ce qu’il devient convenu d’appeler la chimie bio-inorganique.Les moyens expérimentaux et les concepts de la chimie de coordination se révèlent enfin d’une grande efficacité pour l’obtention de composés présentant dans l’état solide des interactions étendues entre cations et, par voie de conséquence, des propriétés physiques, magnétiques ou de conductivité électrique souvent remarquables. L’étude de tels composés va apporter une contribution notable à la physique des matériaux.En définitive les applications pratiques de la chimie de coordination telle qu’elle vient d’être définie sont très nombreuses et, à côté des plus traditionnelles (chimie analytique, chimie des colorants métallifères, hydrométallurgie, tannage des peaux, galvanoplastie, photographie), il faut aujourd’hui mettre en avant ses contributions à la catalyse homogène et à la chimie bio-inorganique.
Les complexes de ligands hydrocarbonés cycliquesDans ce domaine, le ferrocène Fe(C5H5)2 est certainement remarquable à la fois par sa stabilité (il n’est pas détruit à 500 0C) et par sa structure (fig. 7). Dans le motif C5H5Fe, les cinq atomes de carbone de l’anion cyclopentadiényle sont également liés à l’atome métallique et forment une couronne possédant un axe de symétrie d’ordre cinq. Ce motif se retrouve dans de nombreux dérivés appelés parfois «composés sandwich», structuralement analogues au ferrocène, les métallocènes M(C5H5)2, avec M = Ni(II), Cr(II), Co(II), etc., mais aussi dans des dérivés pseudotétraédriques comme (C5H5)2TiCl2 ou (C5H5)2WH2. Par ailleurs, l’anion cyclopentadiényle n’est pas le seul qui puisse être lié symétriquement à un atome métallique; les espèces cycliques C3R3+, C4H42-, C6H6, C7H7+ , C8H82- et leurs dérivés qui, comme C5H5- , possèdent un nombre d’électrons susceptible de leur conférer l’aromaticité, forment également des complexes remarquables dont le prototype est le chrome dibenzène Cr(C6H6)2. On pourra noter de plus que certains anions dérivés du bore, en forme de nid , tel B9C2H11- , conduisent à des composés de type voisin.Enfin, il a été possible de construire des édifices (C5H5)3M2 dans lesquels les trois cycles cyclopentadiényle superposés emprisonnent deux à deux, entre leurs plans parallèles, chacun des deux atomes métalliques placés sur l’axe commun de symétrie de l’ensemble.3. États d’oxydation, coordinences et ligands non usuelsLa chimie de coordination a puisé une part importante de sa dynamique dans la recherche des moyens propres à stabiliser des états d’oxydation particuliers et des états de coordinences non usuelles.Une partie de ce qui précède illustre les très nombreuses possibilités de stabilisation d’états d’oxydation faibles. En ce qui concerne les états d’oxydation élevés des métaux, on sait qu’ils sont souvent stabilisés à l’état d’anions ou polyanions minéraux qu’ils forment avec les ligands 2- et F - . Ils se rencontrent également dans des complexes dérivés de macrocycles azotés [cas de Fe(IV), Mn(IV), Ni(III)...] et de ligands soufrés (dithiocarbamate, dithiolate en particulier). On attribue également à ces états d’oxydation élevés un rôle important en milieu biologique où ils forment des complexes avec certains peptides et polypeptides.L’obtention de complexes présentant des états de coordinence non usuels est facilitée par l’emploi de ligands contraignants, susceptibles d’imposer un environnement particulier à l’ion métallique. Les porphyrines, ligands tétradentates rigides imposant une structure de plan carré, ainsi que certains multidentates [tel P(C6H4N 漣 CH=NH)3, par exemple, qui impose la géométrie de prisme droit] illustrent cet aspect de la stabilisation des complexes.Certaines molécules usuelles simples, dès lors qu’il fut montré qu’elles peuvent jouer un rôle de ligand, ont conduit au développement de larges secteurs de la chimie de coordination. L’un de ceux-ci concerne les complexes de l’azote moléculaire qui jouent un rôle important – de modèles – dans notre compréhension de la fixation de l’azote atmosphérique par les plantes. On en connaît maintenant des exemples pour la plupart des métaux de transition et les propriétés observées pour certains d’entre eux, tel le complexe du molybdène et du bis(diphénylphosphine)éthane, Mo(N2)2(dppe)2, permettent de fonder des espoirs quant à la mise au point de méthodes de synthèse directe de produits azotés intéressants.Les complexes de l’oxygène moléculaire sont également très étudiés, qu’il s’agisse de composés mononucléaires symétriques, dans lesquels les deux atomes d’oxygène sont équidistants du métal, de complexes mononucléaires dans lesquels un seul des atomes d’oxygène est lié au métal, ou qu’il s’agisse enfin de composés dinucléaires dans lesquels l’oxygène sert de pont entre les atomes métalliques. Leur étude apporte des éclaircissements sur le fonctionnement des porteurs d’oxygène (hémoglobine, hémocyanine), d’une part, des oxygénases, des oxydases et des oxygénations catalytiques, d’autre part.Les complexes polynucléairesHier encore simples cas particuliers sans grande portée générale, les complexes polynucléaires des métaux de transition sont l’objet aujourd’hui d’un intérêt considérable, principalement dans le cas où ils font apparaître des liaisons entre atomes métalliques. Dans ce cas précis, on peut distinguer deux catégories de composés, à savoir:– les complexes comportant une (ou plusieurs) liaison métal-métal à deux centres métalliques identiques ou différents; les cas les plus simples de tels complexes dimétalliques sont apparentés à celui de Co2(CO)8 (fig. 2) par le fait que la liaison métal-métal y assure seule l’assemblage de deux motifs monométalliques; dans certains composés, au contraire, les ligands en pont entre les atomes métalliques (tels Cl- , OR- , SR - , PR2- ou RC2- ) viennent renforcer cet assemblage; enfin, des liaisons métal-métal multiples apparaissent pour certaines configurations électroniques relativement pauvres en électrons; on admet ainsi, dans le cas de l’octachlorodimolybdate Mo2Cl84- , dans lequel la distance Mo-Mo est très courte, l’existence d’une liaison quadruple;– les complexes contenant au moins trois atomes métalliques, identiques ou différents, souvent disposés suivant des arrangements polygonaux réguliers (fig. 8); dans ces clusters , les électrons de liaison sont délocalisés sur l’ensemble des atomes métalliques, par ailleurs dans un état d’oxydation formel souvent nul; l’ensemble n’est donc pas sans évoquer le métal massif; les métaux de transition les plus aptes à la synthèse de clusters sont ceux qui se situent sur la droite du tableau périodique; le panorama des structures offert actuellement est extrêmement varié, qu’il s’agisse de composés homo-ou même hétérométalliques, et on connaît maintenant des cas de haute nucléarité (18 atomes de platine par exemple); enfin, certains de ces clusters contiennent, insérés dans le polyèdre métallique, divers petits atomes dans des états de coordinence très inhabituels en chimie moléculaire (fig. 9).Un regain d’intérêt semble enfin se porter sur les polyanions ; ces édifices polymétalliques ne sont en fait pas des clusters dans la mesure où ils ne font pas intervenir d’interaction métal-métal significative sur le plan structural. Résultant le plus souvent de condensations réalisées à partir des ions oxo M4n - (M = VV, NbV, TaV, MoVI, WVI), ils possèdent diverses stœchiométries, tel Nb6198-, que l’on peut proposer comme exemple simple d’isopolyanion, ou (Co2W1242)8- , exemple d’hétéropolyanion. Dans la plupart des cas, ces ions sont formés d’octaèdres M6 partageant sommet et arêtes, à l’exclusion des faces (fig. 10). On peut noter enfin, pour certains d’entre eux, des possibilités de réduction multiélectronique conduisant à des composés à valence mixte.4. Développements récents des méthodes d’études expérimentales et théoriquesLes synthèsesUne grande partie des progrès de la chimie des complexes, en particulier dans le domaine des composés organométalliques, repose sur le développement de méthodes synthétiques fines, tel l’emploi des milieux rigoureusement inertes et celui des basses températures.Les méthodes de séparation sont également d’une grande importance, en particulier les méthodes de chromatographie sur plaque et de chromatographie liquide sous pression. Une mention particulière doit être faite des méthodes de synthèse électrochimique, photochimique, et surtout de la vaporisation d’atomes métalliques. Cette dernière technique, qui consiste à faire réagir directement la vapeur métallique avec des substrats, des ligands divers, a permis de préparer des espèces longtemps inaccessibles, par exemple Ti(C6H6)2.Analyse structuraleLa généralisation des méthodes d’analyse cristallographique, au moyen notamment des diffractomètres automatiques, rend possible la détermination directe des structures moléculaires, qui constitue dans certains domaines des informations strictement irremplaçables (par exemple dans le domaine des clusters). Des techniques encore plus fines combinant la diffraction des rayons X et celle des neutrons autorisent la détermination directe des densités électroniques, en particulier au sein même des liaisons chimiques.Une remarque s’impose qui concerne l’utilisation des méthodes physiques d’investigation. La chimie de coordination est en effet pour une large part – ce qui la différencie largement de la chimie organique classique dans le domaine des déterminations structurales – une chimie des édifices moléculaires de haute symétrie centrée. Et cela confère un rôle essentiel à la théorie des groupes dont l’usage s’est donc imposé aussi bien en spectrographie infrarouge et Raman qu’en spectroscopie électronique ou de résonance magnétique nucléaire ou électronique.Les méthodes d’approche théoriqueLes approches qualitatives, issues de ce qu’il fut convenu d’appeler «théorie des liaisons de valence dirigée», «théorie du champ cristallin» puis «théorie des orbitales moléculaires», s’intègrent maintenant complètement dans un modèle plus large, celui du «champ des ligands» dont elles représentent en quelque sorte des étapes successives d’approximation. Cela résulte directement du fait que chacune de ces approches devait tenir compte de la symétrie de la molécule qui est en fait leur facteur commun. On peut noter également le développement du modèle dit de recouvrement angulaire, qui fournit une évaluation commode de l’énergie de stabilisation liée à la disposition des ligands autour du métal suivant une certaine géométrie. L’effort est maintenant porté sur la mise au point de méthodes de calcul applicables aux atomes lourds des seconde et troisième séries de métaux de transition.5. Réactivité et catalyseLe perfectionnement des concepts classiquesLes réactions de substitution nucléophile du premier et du deuxième ordre S1 et S2 sont étudiées jusqu’au stade le plus intime de leur mécanisme. De plus, des acquisitions importantes dans le domaine de la réactivité ont résulté de l’introduction de la notion de non-rigidité des structures complexes elles-mêmes.La pseudo-rotation dite de Berry permet d’expliquer l’équivalence apparente des ligands axiaux et équatoriaux d’un complexe de structure bipyramidale triangulaire par une déformation légère conduisant à un intermédiaire de structure pyramidale à base carrée [cf. DYNAMIQUE MOLÉCULAIRE].Les réactions fondamentales des complexes organométalliquesCertaines réactions des complexes ont suscité tout particulièrement l’intérêt dans la mesure où elles mettent en évidence la capacité du métal central à modifier profondément l’état structural des réactifs pénétrant la sphère de coordination. Le tableau 2 présente quelques-unes de ces réactions.Ces processus, qui font souvent jouer un rôle de gabarit au métal central du complexe, sont parties intégrantes des mécanismes des réactions catalysées par les complexes des métaux de transition (cf. CATALYSE – catalyse homogène).6. PerspectivesIl est certain que la catalyse homogène organique se place maintenant au tout premier rang des applications de la chimie de coordination (cf. CATALYSE – catalyse homogène). Le domaine des complexes de coordination d’intérêt biologique (on pensera en premier lieu aux chlorophylles et à l’hémoglobine) est également aujourd’hui une puissante motivation pour les spécialistes de la chimie de coordination. De plus, la découverte récente des propriétés antitumorales de certains complexes, en particulier de complexes dérivés du platine, semble marquer le renouveau d’une pharmacopée plus «minérale». Ces domaines relèvent de ce qu’il devient convenu d’appeler la chimie bio-inorganique.Les moyens expérimentaux et les concepts de la chimie de coordination se révèlent enfin d’une grande efficacité pour l’obtention de composés présentant dans l’état solide des interactions étendues entre cations et, par voie de conséquence, des propriétés physiques, magnétiques ou de conductivité électrique souvent remarquables. L’étude de tels composés va apporter une contribution notable à la physique des matériaux.En définitive les applications pratiques de la chimie de coordination telle qu’elle vient d’être définie sont très nombreuses et, à côté des plus traditionnelles (chimie analytique, chimie des colorants métallifères, hydrométallurgie, tannage des peaux, galvanoplastie, photographie), il faut aujourd’hui mettre en avant ses contributions à la catalyse homogène et à la chimie bio-inorganique.
Encyclopédie Universelle. 2012.